Machine Learning Dramatically Streamlines Search for More Efficient Chemical Reactions
Advance by SLAC, Stanford Researchers Greatly Reduces Time Needed to Analyze Complex Reactions, Including Catalysis
By Glennda Chui
Even a simple chemical reaction can be surprisingly complicated. That’s especially true for reactions involving catalysts, which speed up the chemistry that makes fuel, fertilizer and other industrial goods. In theory, a catalytic reaction may follow thousands of possible paths, and it can take years to identify which one it actually takes so scientists can tweak it and make it more efficient.
Now researchers at the Department of Energy’s SLAC National Accelerator Laboratory and Stanford University have taken a big step toward cutting through this thicket of possibilities. They used machine learning – a form of artificial intelligence – to prune away the least likely reaction paths, so they can concentrate their analysis on the few that remain and save a lot of time and effort.
The method will work for a wide variety of complex chemical reactions and should dramatically speed the development of new catalysts, the team reported in Nature Communications.
'A Daunting Task'
“Designing a novel catalyst to speed a chemical reaction is a very daunting task,” said Thomas Bligaard, a staff scientist at the SUNCAT Center for Interface Science and Catalysis, a joint SLAC/Stanford institute where the research took place. “There’s a huge amount of experimental work that normally goes into it.”
For instance, he said, finding a catalyst that turns nitrogen from the air into ammonia – considered one of the most important developments of the 20th century because it made the large-scale production of fertilizer possible, helping to launch the Green Revolution – took decades of testing various reactions one by one.
Even today, with the help of supercomputer simulations that predict the results of reactions by applying theoretical models to huge databases on the behavior of chemicals and catalysts, the search can take years, because until now it has relied largely on human intuition to pick possible winners out of the many available reaction paths.
“We need to know what the reaction is, and what are the most difficult steps along the reaction path, in order to even think about making a better catalyst,” said Jens Nørskov, a professor at SLAC and Stanford and director of SUNCAT.
“We also need to know whether the reaction makes only the product we want or if it also makes undesirable byproducts. We’ve basically been making reasonable assumptions about these things, and we really need a systematic theory to guide us.”
Trading Human Intuition for Machine Learning
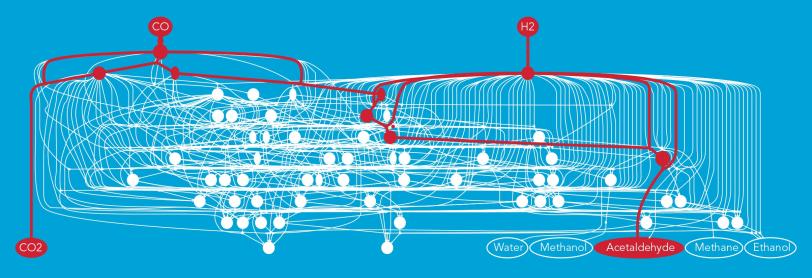
For this study, the team looked at a reaction that turns syngas, a combination of carbon monoxide and hydrogen, into fuels and industrial chemicals. The syngas flows over the surface of a rhodium catalyst, which like all catalysts is not consumed in the process and can be used over and over. This triggers chemical reactions that can produce a number of possible end products, such as ethanol, methane or acetaldehyde.
“In this case there are thousands of possible reaction pathways – an infinite number, really – with hundreds of intermediate steps,” said Zachary Ulissi, a postdoctoral researcher at SUNCAT. “Usually what would happen is that a graduate student or postdoctoral researcher would go through them one at a time, using their intuition to pick what they think are the most likely paths. This can take years.”
The new method ditches intuition in favor of machine learning, where a computer uses a set of problem-solving rules to learn patterns from large amounts of data and then predict similar patterns in new data. It’s a behind-the-scenes tool in an increasing number of technologies, from self-driving cars to fraud detection and online purchase recommendations.
Rapid Weeding
The data used in this process came from past studies of chemicals and their properties, including calculations that predict the bond energies between atoms based on principles of quantum mechanics. The researchers were especially interested in two factors that determine how easily a catalytic reaction proceeds: How strongly the reacting chemicals bond to the surface of the catalyst and which steps in the reaction present the most significant barriers to going forward. These are known as rate-limiting steps.
A reaction will seek out the path that takes the least energy, Ulissi explained, much like a highway designer will choose a route between mountains rather than waste time looking for an efficient way to go over the top of a peak. With machine learning the researchers were able to analyze the reaction pathways over and over, each time eliminating the least likely paths and fine-tuning the search strategy for the next round.
Once everything was set up, Ulissi said, “It only took seconds or minutes to weed out the paths that were not interesting. In the end there were only about 10 reaction barriers that were important.” The new method, he said, has the potential to reduce the time needed to identify a reaction pathway from years to months.
Andrew Medford, a former SUNCAT graduate student who is now an assistant professor at the Georgia Institute of Technology, also contributed to this research, which was funded by the DOE Office of Science.
Citation: Z. Ulissi et al., Nature Communications, 6 March 2017 (10.1038/ncomms14621)
For questions or comments, contact the SLAC Office of Communications at communications@slac.stanford.edu.
SLAC is a multi-program laboratory exploring frontier questions in photon science, astrophysics, particle physics and accelerator research. Located in Menlo Park, Calif., SLAC is operated by Stanford University for the U.S. Department of Energy's Office of Science.
SLAC National Accelerator Laboratory is supported by the Office of Science of the U.S. Department of Energy. The Office of Science is the single largest supporter of basic research in the physical sciences in the United States, and is working to address some of the most pressing challenges of our time. For more information, please visit science.energy.gov.
Related stories